1. Getting started with Escher visualizations¶
1.1. Introduction¶
Escher is web application for visualizing metabolic pathway maps.
Three key features make Escher a uniquely effective tool for pathway visualization. First, users can rapidly design new pathway maps. Escher provides pathway suggestions based on user data and genome-scale models, so users can draw pathways in a semi-automated way. Second, users can visualize data related to genes or proteins on the associated reactions and path- ways, using rules that define which enzymes catalyze each reaction. Thus, users can identify trends in common genomic data types (e.g. RNA-Seq, proteomics, ChIP)—in conjunction with metabolite- and reaction-oriented data types (e.g. metabolomics, fluxomics). Third, Escher harnesses the strengths of web technologies (SVG, D3, developer tools) so that visualizations can be rapidly adapted, extended, shared, and embedded.
The rest of this guide will introduce the Escher user interface and the major features of Escher.
1.2. The launch page¶
When you open the Escher website, you will see a launch page that looks like this:
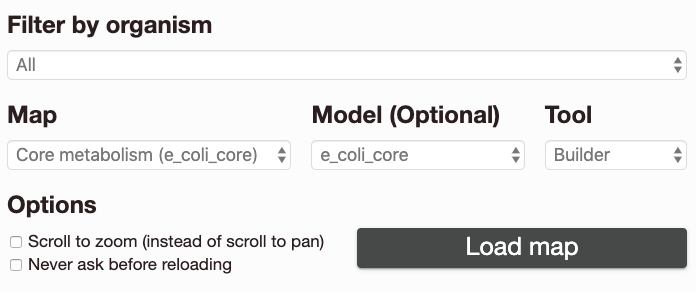
The options on the launch page are:
Filter by organism: Choose an organism to filter the Maps and Models.
Map: Choose a pre-built map, or start from scratch with an empty builder by choosing None. In parentheses next to the map name, you will see the name of the model that was used to build this map.
Model: (Optional) If you choose a COBRA model to load, then you can add new reactions to the pathway map. You can also load your own model later, after you launch the tool. For an explanation of maps, models, and COBRA, see Escher, COBRA, and COBRApy.
Tool:
The Viewer allows you to pan and zoom the map, and to visualize data for reactions, genes, and metabolites.
The Builder, in addition to the Viewer features, allows you to add reactions, move and rotate existing reactions, add text annotations, and adjust the map canvas.
Options:
Scroll to zoom (instead of scroll to pan): Determines the effect of using the mouse’s scroll wheel over the map.
Never ask before reloading: If this is checked, then you will not be warned before leaving the page, even if you have unsaved changes.
Choose Load map to open the Escher viewer or builder in a new tab, and prepare to be delighted by your very own pathway map.
1.3. The menu bar¶
Once you have loaded an Escher map, you will see a menu bar along the top of the screen. Click the question mark to bring up the Escher documentation:

1.4. Loading and saving maps¶
Using the map menu, you can load and save maps at any time:
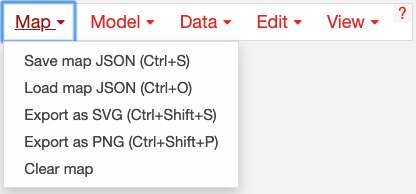
Click Save map JSON to save the Escher map as a JSON file, which is the standard file representing an Escher map.
NOTE: The JSON file does NOT save any datasets you have loaded. This may be changed in a future version of Escher.
NOTE 2: In Safari, saving files from Escher works a little differently than
in the other major browsers. After clicking Save map JSON, Safari will load
a new tab with the raw content of the Escher map file. To save the file, choose
File>Save As… from the Safari menu. A new dialog appears; in the dropdown menu
near the bottom, select Page Source (rather than Web Archive), give your file an
appropriate name (e.g. map.json for an Escher map or map.svg for an SVG
export), and click Save.
Later, you can can load a JSON file to view and edit a map by clicking Load map JSON.
Click Export as SVG to generate a SVG file for editing in tools like Adobe Illustrator and Inkscape. This is the best way to generate figures for presentations and publications. Unlike a JSON file, a SVG file maintains the data visualizations on the Escher map. However, you cannot load SVG files into Escher after you generate them.
Click Export as PNG to generate a `PNG`_ file for sharing finished visualizations quickly (e.g. in PowerPoint).
Click Clear Map to empty the whole map, leaving a blank canvas. NOTE: You cannot undo Clear Map.
1.5. Loading models¶
Use the model menu to manage the COBRA model loaded in Escher:
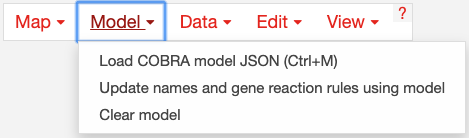
Choose Load COBRA model JSON to open a COBRA model. Read more about COBRA models in Escher, COBRA, and COBRApy. For examples of using Escher with COBRApy, see the Escher Python tutorial.
Once you have loaded a COBRA model, there may be inconsistencies between the content in the map and the model (e.g. reaction IDs, descriptive names and gene reaction rules). You click Update names and gene reaction rules using model to find matching reactions and metabolites between the map and the model (based on their IDs) and then apply the names and gene reaction rules from the model to the map. The reactions that do not match will be highlighted in red. (This can be turned off again in the settings menu by deselecting Highlight reactions not in model.) More advice on building maps is available in contribute_maps.
Click Clear Model to clear the current model.
1.6. Loading reaction, gene, and metabolite data¶
Datasets can be loaded as CSV files or JSON files, using the Data Menu.
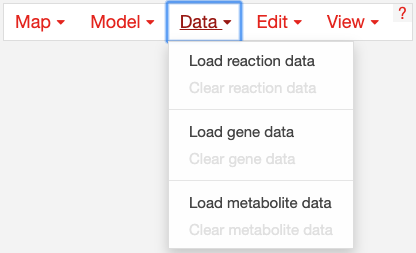
In Escher, reaction and gene datasets are visualized by changing the color, thickness, and labels of reaction arrows. Metabolite datasets are visualized by changing the color, size, and labels of metabolite circles. The specific visual styles can be modified in the Settings menu. When data is not present for a specific reaction, gene, or metabolite, then the text label will say ‘nd’ which means ‘no data.’
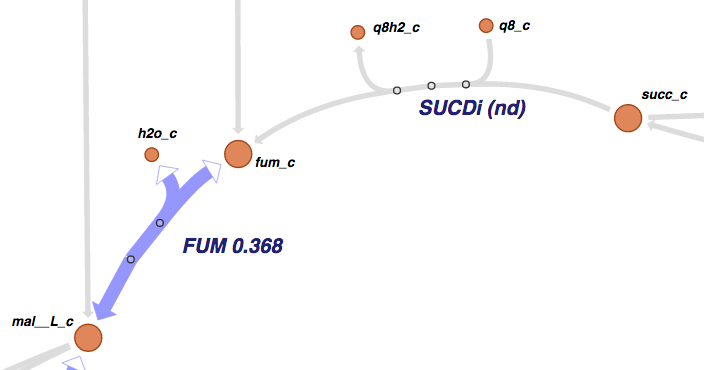
1.6.1. Example data files¶
It is often easiest to learn by example, so here are some example datasets that work with Escher maps for the Escherichia coli model iJO1366:
Reaction data
S3_iJO1366_anaerobic_FBA_flux.json: FBA flux simulation data for iJO1366 as JSON.
reaction_data_iJO1366.json: A JSON file with one dataset of fluxes.
reaction_data_diff_iJO1366.json: A JSON file with two dataset of fluxes.
Metabolite data
S4_McCloskey2013_aerobic_metabolomics.csv: Aerobic metabolomics for E. coli as CSV.
metabolite_data_iJO1366.json: A JSON file with one dataset of metabolite concentrations.
metabolite_data_diff_iJO1366.json: A JSON file with two datasets of metabolite concentrations.
Gene data
S6_RNA-seq_aerobic_to_anaerobic.csv: Comparison of two gene datasets (RNA-seq) as CSV.
gene_data_names_iJO1366.json: A single gene dataset using descriptive (gene) names for identifiers as JSON.
1.6.2. Creating data files as CSV and JSON¶
CSV files should have 1 header row, 1 ID column, and either 1 or 2 columns for data values. The ID column can contain BiGG IDs or descriptive names for the reactions, metabolites, or genes in the dataset. Here is an example with a single data value columns:
ID,time 0sec
glc__D_c,5.4
g6p__D_c,2.3
Which might look like this is Excel:
ID |
time 0sec |
|---|---|
glc__D_c |
5.4 |
g6p_c |
2.3 |
If two datasets are provided, then the Escher map will display the difference between the datasets. In the Settings menu, the Comparison setting allows you to choose between comparison functions (Fold Change, Log2(Fold Change), and Difference). With two datasets, the CSV file looks like this:
ID |
time 0sec |
time 5s |
|---|---|---|
glc__D_c |
5.4 |
10.2 |
g6p_c |
2.3 |
8.1 |
Data can also be loaded from a JSON file. This Python code snippet provides an example of generating the proper format for single reaction data values and for reaction data comparisons:
import json
# save a single flux vector as JSON
flux_dictionary = {'glc__D_c': 5.4, 'g6p_c': 2.3}
with open('out.json', 'w') as f:
json.dump(flux_dictionary, f)
# save a flux comparison as JSON
flux_comp = [{'glc__D_c': 5.4, 'g6p_c': 2.3}, {'glc__D_c': 10.2, 'g6p_c': 8.1}]
with open('out_comp.json', 'w') as f:
json.dump(flux_comp, f)
1.6.3. Gene data and gene reaction rules¶
Escher uses gene reaction rules to connect gene data to the reactions on a metabolic pathway. You can see these gene reaction rules on the map by selecting Show gene reaction rules in the Settings menu.
Gene reaction rules show the genes whose gene products are required to catalyze a reaction. Gene are connected using AND and OR rules. AND rules are used when two genes are required for enzymatic activity, e.g. they are members of a protein complex. OR rules are used when either gene can catalyze the enzymatic activity, e.g. they are isozymes.
With OR rules, Escher will take the sum of the data values for each gene. With AND rules, Escher will either take the mean (the default) or the minimum of the components. The AND behavior (mean vs. minimum) is defined in the Settings menu.
1.7. Editing and building¶
The Edit menu gives you access to function for editing the map:
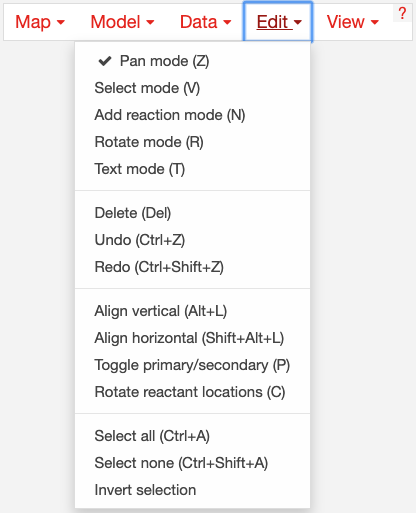
Escher has five major modes, and you can switch between those modes using the buttons in the Edit menu, or using the buttons in the button bar on the left of the screen.
Pan mode: Drag the canvas to pan the map. You can also use the mouse scroll wheel or trackpad scroll function (drag with 2 fingers) to pan the map (or to zoom if you selected Scroll to zoom in the settings).
Select mode: Select nodes by clicking on them. Shift-click to select multiple nodes, or drag across the canvas to select multiple nodes using the selection brush.
Add reaction mode: If you have loaded a Model, then click on the canvas to see a list of reactions that you can draw on the map. Click on a node to see reactions that connect to that node. In the input box, you can search by reaction ID, metabolite ID, or gene ID (locus tag).
Rotate model: Before entering rotate mode, be sure to select one or more nodes in select mode. Then, after entering rotate mode, drag anywhere on the canvas to rotate the selection. You can also drag the red crosshairs to change the center of the rotation.
Text mode: Use text mode to add text annotations to the map. Click on the canvas to add a new text annotation, or click an existing annotation to edit it. When you are finished, click Enter or Escape to save the changes.
In addition to the editing modes, the Edit menu gives you access to the following commands:
Delete: Delete the currently selected node(s).
Undo: Undo the last action. NOTE: Certain actions, such as editing the canvas, cannot be undone in the current version of Escher.
Redo: Redo the last action that was undone.
Align vertical and Align horizontal: Line up with selected nodes in the given direction. If you have selected entire reactions, Escher will try to intelligently maintain the shape of the reactions.
Toggle primary/secondary node: In Escher, each metabolite node is either a primary node or a secondary node. Primary nodes are larger, and secondary nodes can be hidden in the Settings menu. Use this command to toggle the currently selected node(s) between primary and secondary.
Rotate reactant locations: When you draw a new reaction in Escher, this command will rotate the new reactants so that a new reactant is primary and centered. This command is extremely useful when you are drawing a long pathway and you want to quickly switch which metabolite to “follow”, e.g. make sure you are following the carbon-containing metabolites.
If you are confused, then try drawing a new pathway and hitting the “c” key to see the reactants rotate.
Select all: Select all nodes.
Select none: Unselect all nodes.
Invert selection: Select all the nodes that are currently unselected. This feature is very useful when you want to keep just one part of the map. Simply drag to select the part you want to keep, call the Invert selection command, then call the Delete command.
1.8. Editing the canvas¶
A somewhat non-obvious feature of Escher is that you can edit the canvas by dragging the canvas edges. This is possible in Pan mode and Select mode.
1.9. View options¶
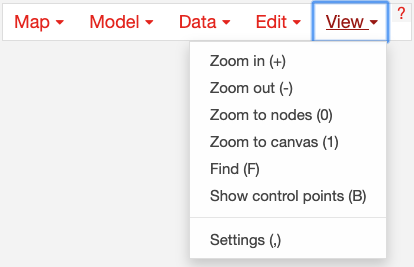
Zoom in: Zoom in to the map.
Zoom out: Zoom out of the map.
Zoom to nodes: Zoom to see all the nodes on the map.
Zoom to canvas: Zoom to see the entire canvas.
Find: Search for a reaction, metabolite, or gene on the map.
Show control points: Show the control points; you can drag these red and blue circle to adjust the shapes of the reactions curves.
Settings: Open the Settings menu.

1.11. Settings¶
1.11.1. View and build options¶

Identifiers: Choose whether to show BiGG IDs or descriptive names for reactions, metabolites, and genes.
Scroll to zoom: If checked, then the scroll wheel and trackpad will control zoom rather than pan.
Hide secondary metabolites: This will simplify the map by hiding all secondary metabolites and the paths connected to them.
Show gene reaction rules: Show the gene reaction rules below the reaction labels, even gene data is not loaded.
Hide reaction, gene, and metabolite labels: Another option to visually simplify the map, this will hide all labels that are not text annotations.
Allow duplicate reactions: By default, duplicate reactions are hidden in the add reaction dropdown menu. However, you can turn this option on to show the duplicate reactions.
Highlight reactions not in model: Highlight in red any reactions that are on the map but are not in the model. This is useful when you are adapting a map from one model for use with another model
Use 3D transform for responsive panning and zooming: If true, then use CSS3 3D transforms to speed up panning and zooming.
Show tooltips: Determines over which elements tooltips will display for reactions, metabolites, and genes
1.11.2. Reaction data settings¶
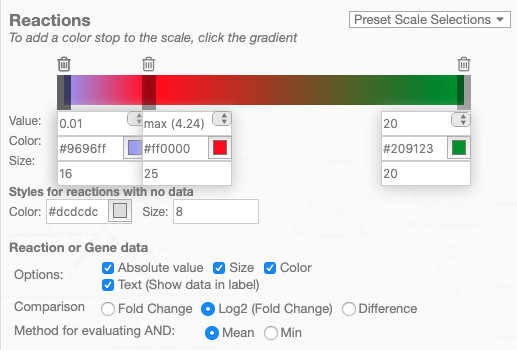
When reaction or gene data is loaded, this section can be used to change visual settings for reactions.
The color bar has individual control points, and you can drag the control points (execpt min and max) left and right to change their values. Alternatively, you can use the dropdown menu (next to the word median in the figure above), to attach a control point to a statistical measure (mean, median, first quartile (Q1), or third quartile (Q3)). This lets you choose a color scale that will adapt to your dataset.
For each control point, you can choose a color by entering a CSS-style color (e.g. red, #ff0000, rgba(20, 20, 255, 0.5), and you can choose a size that will scale the thickness of reactions.
There are also color and size options for reactions that do not have any data value.
The Preset Scale Selections menu provides some built-in color scales that will modify the scale for you with new control points, colors, and sizes.
Finally, there are a few on/off settings for the loaded reaction or gene dataset:
Options: These determine how to visualize the datasets. Check Absolute value to color and size reactions by the absolute value of each data value. The Size, Color, and Text options can be unselected to turn off sizing, coloring, and data values in reaction labels individually.
Comparison: Determines the comparison algorithm to use when two datasets are loaded.
Method for evaluating AND: Determines the method that will be used to reconcile AND statements in gene reaction rules when there is gene data loaded. See Gene data and gene reaction rules for more details.
1.11.3. Metabolite data settings¶
The data settings for metabolite data are analagous to those for reaction data. The only difference is that size now refers to the size of the metabolite circles.